The arrival of a newborn marks a period of intensive physiological transition. One of the most critical milestones in the first hours of life is the passage of meconium—the dark, viscous material that fills the infant's intestines during gestation. While 99 percent of full-term infants pass this first stool within 48 hours, a small subset of newborns fails to do so due to a mechanical obstruction known as meconium ileus. This condition occurs when the meconium becomes excessively thick, dry, and sticky, effectively sealing the terminal ileum. Understanding the diagnostic pathway for meconium ileus is vital, as it frequently serves as the earliest clinical indicator of Cystic Fibrosis (CF).
The Physiological Basis of Obstruction
Meconium ileus represents more than just a simple blockage; it is a manifestation of abnormal exocrine gland function. In a healthy fetus, intestinal secretions remain fluid enough to allow the meconium to pass through the digestive tract. However, in approximately 15 to 20 percent of infants with Cystic Fibrosis, the lack of functional chloride channels leads to dehydrated, protein-rich intestinal secretions. This results in meconium with the consistency of thick glue or tar, which adheres to the intestinal walls and refuses to move.
This obstruction typically occurs in the terminal ileum, the final section of the small intestine. The bowel segments above the blockage dilate significantly as they attempt to push the material forward, while the segments below remain narrow and unused (microcolon). If the obstruction occurs during fetal life, it can lead to complications such as intestinal volvulus, perforation, or meconium peritonitis, necessitating emergency surgical intervention immediately after birth.
Recognizing the Clinical Presentation
Healthcare providers in the newborn nursery maintain high vigilance for the classic triad of symptoms associated with intestinal obstruction. Early identification allows for the initiation of diagnostic tests before the infant experiences significant metabolic stress or bowel injury. Because the symptoms of meconium ileus often overlap with other neonatal conditions, the clinical history provides the first piece of the diagnostic puzzle.
Radiological Imaging and Contrast Studies
When a clinician suspects meconium ileus, the first diagnostic step involves abdominal radiography. A plain X-ray provides a snapshot of the gas patterns within the infant's gut. In meconium ileus, the X-ray often shows dilated loops of small bowel with a characteristic lack of air-fluid levels, which contrasts with other forms of obstruction. A specific finding known as the Neuhauser sign (a "soap-bubble" appearance) occurs when air mixes with the thick, inspissated meconium in the ileum.
The Contrast Enema: Diagnostic and Therapeutic
If the plain X-ray suggests a low intestinal obstruction, the definitive imaging test is a Contrast Enema (often using a water-soluble, hyperosmolar agent like Gastrografin). This procedure involves gently introducing a specialized dye into the rectum while a radiologist observes the flow via fluoroscopy. This test serves two primary purposes:
First, it identifies the "microcolon," a very narrow, unused colon that confirms the obstruction is located higher up in the small intestine. Second, it can be therapeutic. The hyperosmolar contrast agent draws water into the bowel lumen, which softens the thick meconium and helps it detach from the intestinal wall. In many uncomplicated cases, the contrast enema successfully clears the blockage, avoiding the need for surgery.
The Gold Standard: Sweat Chloride Testing
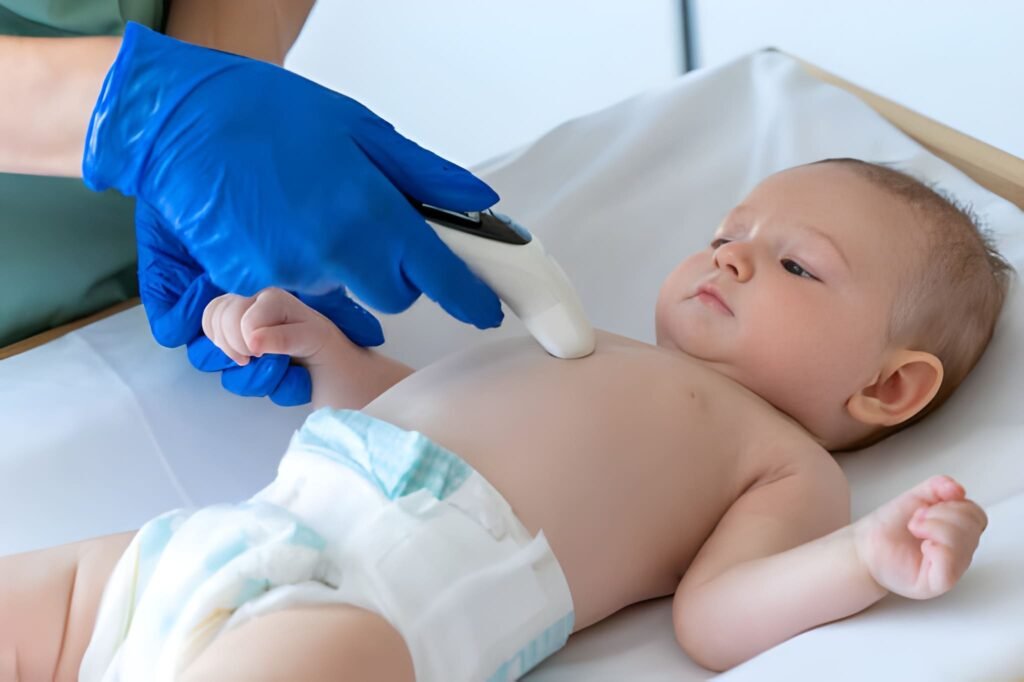
Once the intestinal obstruction is managed or identified, the clinical focus shifts to the underlying cause. The Sweat Chloride Test is the definitive diagnostic test for Cystic Fibrosis. It measures the concentration of salt in the infant's sweat. Because the CFTR protein—which moves chloride in and out of cells—is defective in CF, the sweat becomes excessively salty.
Performing this test on a newborn requires patience, as the infant must produce enough sweat for a valid sample. Technicians apply a chemical called pilocarpine to a small area of the skin and use a mild electric current to stimulate the sweat glands. The sweat is then collected and analyzed. In a newborn, the test is typically performed after the infant is at least 48 hours old and is sufficiently hydrated.
Normal Range: Less than or equal to 29 mmol/L.
Intermediate (Inconclusive): 30 to 59 mmol/L.
Positive (Diagnostic for CF): Greater than or equal to 60 mmol/L.
Example Calculation: If a newborn with meconium ileus yields a sweat chloride result of 72 mmol/L, the probability of Cystic Fibrosis is exceptionally high. Conversely, a result of 15 mmol/L suggests that the ileus may have a different, rarer cause, such as meconium plug syndrome or Hirschsprung disease.
Genetic Analysis and Newborn Screening
In the United States and many other regions, Newborn Screening (NBS) programs include a check for Cystic Fibrosis. This usually involves measuring Immunoreactive Trypsinogen (IRT) from the initial heel-stick blood sample. If the IRT is elevated, the laboratory may automatically perform a DNA analysis to look for the most common CF mutations, such as the Delta F508 mutation.
However, it is vital to remember that a newborn with meconium ileus may show a positive diagnosis even before the NBS results return. If the clinical presentation is strong and the sweat test is positive, genetic testing serves to confirm the specific mutations involved. This information is crucial for family planning and for determining if the infant is a candidate for modern CFTR modulator therapies, which target the specific protein defect based on the individual's genetic profile.
Differential Diagnosis Comparison
Not all failures to pass meconium are caused by meconium ileus. Physicians must differentiate between several neonatal conditions that present with similar symptoms. The following table highlights the key differences between these diagnostic possibilities.
| Condition | Obstruction Site | Radiological Finding | Associated Condition |
|---|---|---|---|
| Meconium Ileus | Terminal Ileum | Microcolon; Soap-bubble sign | Cystic Fibrosis (80-90%) |
| Hirschsprung Disease | Distal Colon/Rectum | Transition zone; dilated colon | Nerve cell deficiency |
| Meconium Plug Syndrome | Colon | Movable plugs of meconium | Maternal diabetes; prematurity |
| Ileal Atresia | Small Intestine | Multiple air-fluid levels | Structural bowel defect |
Socioeconomic Context and Access to Testing
In the United States, the availability of high-level neonatal intensive care units (NICUs) ensures that most cases of meconium ileus are diagnosed and treated rapidly. However, disparities in access to specialized pediatric surgeons and genetic counselors can affect long-term outcomes. Furthermore, while the sweat test is relatively inexpensive, the cost of ongoing Cystic Fibrosis management—including medications, chest physiotherapy, and specialized nutrition—can place a significant burden on families. Comprehensive insurance coverage and state-funded programs play a critical role in supporting these families from the moment of the newborn diagnosis through adulthood.
Management and Long-Term Outlook
The initial diagnosis of meconium ileus is often a traumatic experience for new parents. However, with early identification and a multi-disciplinary approach involving neonatologists, pediatric surgeons, and CF specialists, the prognosis has improved dramatically over the last few decades. For infants whose ileus is resolved non-surgically via contrast enema, the focus quickly shifts to optimizing nutrition and preventing respiratory complications associated with CF. For those who require surgery, modern techniques like the creation of an ileostomy allow the bowel to rest and heal before being reconnected later in infancy.
Early diagnosis via meconium ileus provides a "head start" on the management of Cystic Fibrosis. By beginning treatment before the infant develops chronic lung infections or significant pancreatic insufficiency, medical teams can significantly improve the quality of life and life expectancy for these children. The diagnostic tests—from the initial X-ray to the final genetic confirmation—are the first steps in a lifelong journey toward health and stability.